
Vilhelm Bohr
Vilhelm Bohr is primarily interested in the processes of DNA maintenance, mitochondrial biology and energy metabolism, and how these processes dynamically influence one another with implication on the aging process and age-associated neurodegeneration including Alzheimer disease (AD).
Background
Our genome is constantly damaged at considerably high numbers per cell per day by spontaneous hydrolytic decay and by various endogenous and environmental agents. In addition, nucleotide insertion errors during DNA replication and the process of DNA repair itself constitute substantial sources of genome modifications. The preservation of the chemical structure and the original nucleotide sequence of the genome are essential for life. Thus, mechanisms have evolved in all living organisms to repair DNA lesions and to maintain genome stability and integrity.
Aging is broadly defined as a progressive decline in organismal function over time associated with an increasing risk of disease and death. At the molecular level, the aging process is commonly thought to be a consequence of a lifelong accumulation of stochastic damage to macromolecules. A substantial body of evidence indicates that DNA damage and mutation (both nuclear and mitochondrial DNA) accumulate with age in multiple human and animal tissues.
Mitochondria are the prominent sites for the production of the main cellular energy substrate ATP and these organelles play a pivotal role in several other key cellular processes. The evolutionary conserved and functionally interconnected systems of nutrition-sensing, cellular energy homeostasis, and genome maintenance, implicate mitochondrial physiology and maintenance. Failure in these pathways can impair mitochondrial function and turnover of damaged mitochondria, hallmarks of many human pathologies and aging. Increasing knowledge of these mechanisms has contributed to the development of pharmacological and dietary intervention strategies with promising effect on lifespan and health-span in animal models and potentially also in humans.
Our group is primarily interested in the processes of DNA maintenance, mitochondrial biology and energy metabolism, and how these processes dynamically influence one another with implication on the aging process and age-associated neurodegeneration including Alzheimer disease (AD).
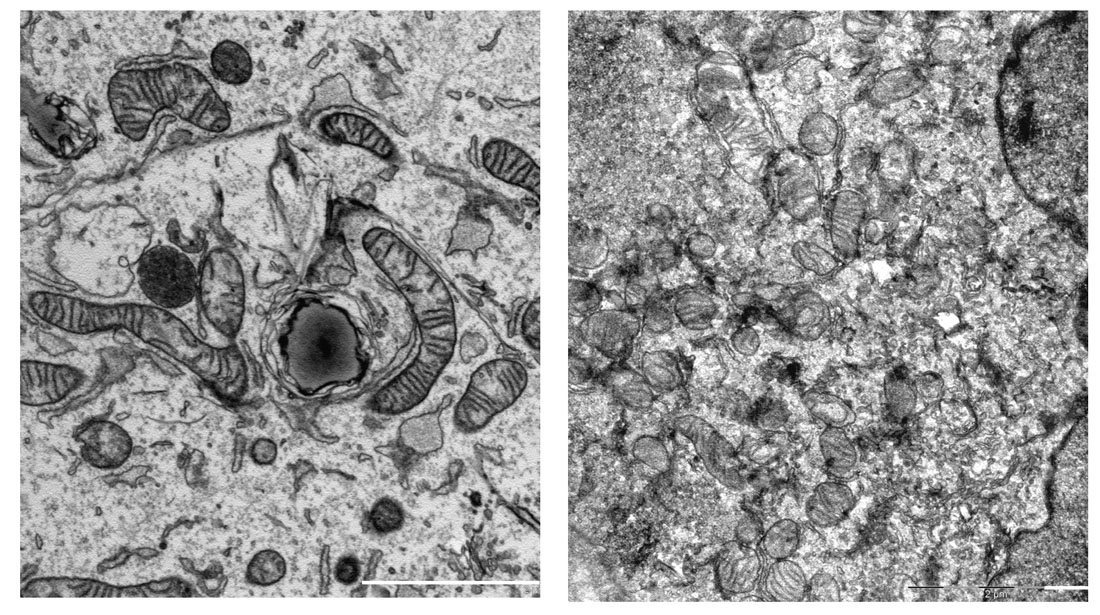
Transmission electron microscopy images showing highly fragmented mitochondria in BER deficient U2OS cells (right) as compared to the control cells (left). Scale bar represents 2 μm. The image was prepared at the Core facility of Integrated Microscopy, University of Copenhagen.
i) We and others have identified nicotinamide adenine dinucleotide (NAD+), as a key cofactor linking nuclear DNA damage to mitochondrial dysfunction and neurodegeneration. Moreover, we have demonstrated that treating animal models of human neurodegenerative diseases including a mouse model of AD, with NAD+ precursors as well as compounds that boost autophagic removal of dysfunctional mitochondria, improve mitochondrial function and reverse key neurological impairment in these animals. The aim of this project is to study the molecular mechanisms linking NAD+ metabolism and the process of autophagy to mitochondrial function and to investigate pharmacological treatments that can improve neurological performance using different C. elegans strains of human neurodegenerative diseases.
ii) Neuropathology of AD is characterized by deposition of amyloid-b peptide plaques and intraneuronal neurofibrillary tangles (NFTs) composed of hyperphosphorylated tau protein, synaptic degeneration and neuronal death. Tau is a microtubule-associated protein important for assembly and stabilization of microtubules and is abundantly present in the central nervous system with predominant activity in the distal portions of axons. Tau has also been detected in the nucleus where it seems to play genome protection and gene expression regulatory functions. The primary objective of this project is to investigate the interplay between DNA damage, mitochondrial stress and tau pathology including abnormal tau phosphorylation, tau oligomers, and tangles. We are using a mouse model of AD (3xTg, Polb -/+), a tau knockout mouse, a mouse model of tau aggregation (THY-Tau22), and human cell lines.
iii) DNA ligation can result in 5´-adenylated (5´-AMP) DNA termini. Aprataxin (APTX) catalyzes direct removal of 5´-AMP adducts from DNA. Mutations in APTX cause an inherited disease characterized by early-onset progressive ataxia with ocular motor apraxia (AOA1). We have reported that APTX localizes to both nucleus and mitochondria and that APTX deficiency renders more mitochondrial DNA to damage than nuclear DNA. In this project, we are studying the effect of APTX deficiency on mitochondrial physiology, and how alterations in mitochondrial biology might be linked to AOA1 pathology, in CRISPR-mediated APTX knock-out human cell lines.
iv) RECQL4 belongs to the RecQ family of helicases also called the guardians of the genome. Mutations in RECQL4 associate with Rothmund-Thomson (RTS), Baller–Gerold (BGS), and RAPADILINO syndromes, rare autosomal recessive disorders characterized by premature aging, genomic instability, and cancer predisposition. RECQL4 is mostly a nuclear protein, but a fraction of the enzyme is present in the cytoplasm and in mitochondria. Increased RECQL4 expression and cytosolic accumulation of the protein has been detected in a number of clinical tumor samples including breast cancer, prostate cancer, and osteosarcoma. Moreover, RAPADILINO RECQL4 mutant accumulates in the cytosol. The aim of this project is to study role of RECQL4 defect on mitochondrial function and cell metabolism and how those processes are linked to diseases related to RECQL4 deficiency using CRISPR-mediated RECQL4 knock-out cells, cells that express RAPADILINO RECQL4, and tissues from a mouse model of RTS. Moreover, using a combination of clinical materials and cell lines, we are studying the role of abnormal cytoplasmic accumulation of RECQL4 in cancer progression.
- Drs. Marie-Christine Galas, and Bart Dermaut, University of Lille, Inserm, France.
- Dr. Joakim Lundeberg, KTH Royal Institute of Technology, Sweden.
- Drs. Tone Tønjum, Hilde Nilsen, and Evandro Fang, University of Oslo, Norway.
- Dr. Tinna Stevnsner, University of Aarhus, Denmark.
- Dr. Lene Juel Rassmussen, University of Copenhagen, Denamark.
- Fang EF., et al (2019) Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer's disease. Nature Neuroscience.
- Zheng J., et al., (2019) Diminished OPA1 expression and impaired mitochondrial morphology and homeostasis in Aprataxin-deficient cells. Nucleic Acids Research
- Fang EF. et al., (2017) Tomatidine enhances lifespan and healthspan in C. elegans through mitophagy induction via the SKN-1/Nrf2 pathway. Scientific Reports.
- Hou Y. et al., (2017) Genome instability in Alzheimer disease. Mechanisms of Ageing and Development.
- Sykora P. et al., (2017) DNA polymerase beta participates in mitochondrial DNA repair. Molecular and Cellular Biology.
- Fang E.F. et al., (2016) NAD+ Replenishment Improves Lifespan and Healthspan in Ataxia Telangiectasia Models via Mitophagy and DNA Repair. Cell Metabolism.
- Akbari M. et al., (2015) The role of DNA base excision repair in brain homeostasis and disease. DNA Repair.
- Akbari M. et al., (2015) Slow mitochondrial repair of 5'-AMP renders mtDNA susceptible to damage in APTX deficient cells. Scientific Reports.
- Maynard S. et al., (2015) Defective mitochondrial respiration, altered dNTP pools and reduced AP endonuclease 1 activity in peripheral blood mononuclear cells of Alzheimer's disease patients. Aging.
- The Nordea Foundation Center for Healthy Aging
- EU Joint Programme-Neurodegenerative Disease Research (JPND)
- Innovation Fund Denmark
- Olav Thon Foundation, Norway
- Novo Nordisk Foundation, Denmark
 Vilhelm Bohr
Vilhelm Bohr
Affiliate Professor in Genome instability and neurodegeneration
vbohr@sund.ku.dk
(+45) 35 32 67 98
National Institutes of Health (USA), bohrv@grc.nia.nih.gov