
Svejstrup Group
We investigate the molecular mechanisms of transcription, and how this essential cellular process interfaces with other DNA-related processes, such a DNA replication and repair.
We study the interface between transcription and other DNA-related processes, such as DNA repair, replication, and recombination.
One of the hallmarks of human disease, including cancer, is the deregulation of gene expression: genes that are supposed to be inactive become activated, or vice versa. Another characteristic of cancer cells is their unstable genomes. They are unable to repair or maintain the integrity of their DNA.
Over the last decade, it has also become increasingly clear that the process of transcription itself entails a significant risk for genome stability. The mechanisms that ensure the safe co-existence of transcription with DNA replication and pathways maintaining genome integrity are still poorly understood.
We seek to understand transcription and the maintenance of genome integrity using a unique combination of biochemical, genetic and cell biological techniques, primarily in human cells.
Basic mechanisms of transcript elongation
Transcription of mRNA-encoding genes underpins all life. Most fundamental processes in cells and organisms are regulated at the level of transcription.
DNA damage in genes can directly give rise to harmful mutations, but it can also obstruct the progress of transcribing RNA polymerase II (RNAPII), thereby blocking gene expression. A repair pathway has evolved that specifically targets lesions that stop RNAPII during its journey across a gene, so-called transcription-coupled nucleotide excision repair (TC-NER). Damage-stalled RNAPII can also be removed by ubiquitylation/degradation, clearing the gene for repair by other means.
While transcription is essential and therefore protected by a variety of mechanisms, it also comes at a cost for genome integrity. For example, high levels of transcription are correlated with breaks at fragile chromosome sites, mutagenesis and elevated levels of DNA recombination.
Research into how the genome-destabilising effects of transcription are minimized is still at a very early stage, but insights into this research area is essential for our understanding of the regulatory mechanisms at play in the interface between transcription and other DNA-related processes, as well as for the understanding of processes underlying genome instability.
Our laboratory studies the mechanisms of transcription, with particular emphasis on transcript elongation in human cells, using a wide range of experimental approaches, ranging from reconstitution biochemistry using purified proteins, over proteomics and genomics, to cell biological and genomic screening techniques.
Transcription-coupled DNA repair
Bulky DNA lesions such as those generated by UV-irradiation block the progress of RNA polymerase II and therefore pose a particular threat to genome integrity and cell survival. Multiple mechanisms have evolved to minimise their detrimental effect.
Transcription-coupled nucleotide excision repair (TC-NER) ensures that the transcribed strand of an active gene is repaired much faster than the non-transcribed strand and the genome in general. Elucidating the mechanism of TC-NER is an important part of understanding the mechanisms of DNA repair.
Research in several laboratories has shown that Cockayne syndrome B (CSB) protein is recruited to damage-stalled RNAPII and is instrumental in recruiting basal nucleotide excision repair (NER) factors, as well as proteins required for subsequent repair-dependent DNA synthesis.
Without CSB, no TC-NER complex is assembled, and the CSA protein is not recruited either. CSA is dispensable for both CSB recruitment and TC-NER complex assembly, but is still absolutely required for TC-NER to take place. CSA is a component of a cullin-based ubiquitin ligase complex, but the functional importance of this activity in TC-NER is not known.
Interestingly, however, we have found that CSB contains a ubiquitin-binding domain (UBD), which is dispensable for repair complex assembly, but essential for activation of the DNA incision. With the discovery that CSB contain a functionally important UBD, it is an obvious possibility that CSA/Cullin-mediated ubiquitylation of a factor in the repair complex is recognized by CSB via its UBD, and that this in turn regulates the repair reaction.
It is a major goal for our research into transcription-coupled repair to understand the biochemical function of the CSA-cullin complex and CSB translocase in human cells.
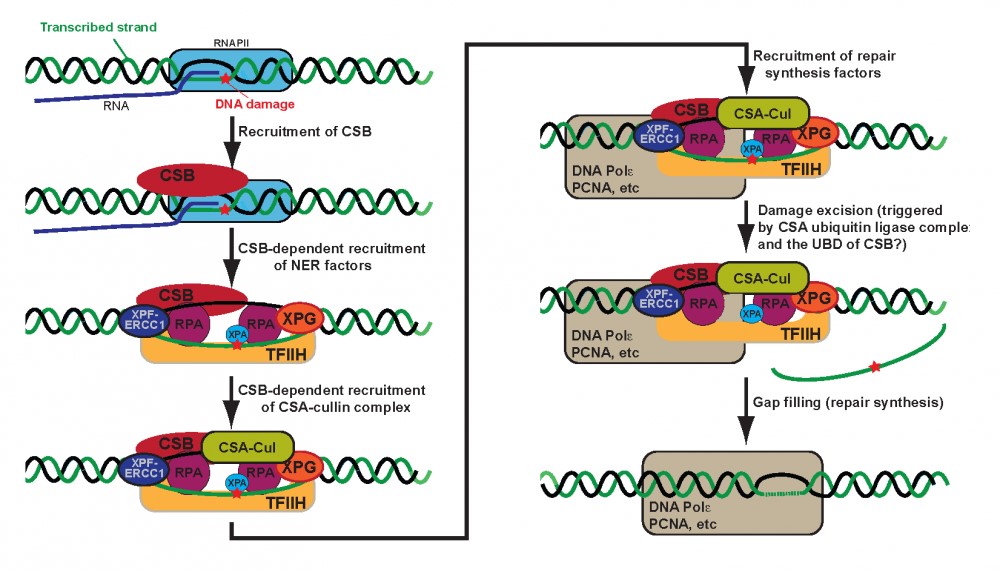
Figure 1. Stepwise model for TC-NER. Note that RNAPII is likely to be present during the whole reaction and to restart transcription after repair, but it has been omitted from the cartoon in later steps for simplicity.
Ubiquitylation of RNA polymerase II and other proteins (short: protein Ubiquitylation)
Damage-stalled RNAPII functions as a molecular ‘beacon’ that triggers transcription-coupled nucleotide excision repair (TC-NER).
On the other hand, if that DNA lesion for some reason cannot be removed by TC-NER, a ‘mechanism of last resort’ ensures that RNAPII is ubiquitylated and degraded by the proteasome, enabling repair by other mechanisms, such as global genomic nucleotide excision repair.
Our lab has contributed significantly to the understanding of this last resort pathway over the last two decades. However, the mechanism and factor requirement of RNAPII ubiquitylation and degradation is still not fully understood.
Likewise, it is increasingly clear that not only RNAPII but also a large number of other factors become ubiquitylated in response to DNA damage, and that this is crucial for correct regulation of the DNA damage response. We have developed powerful methods to analyze ubiquitylation on a proteome-wide scale and used them to map hundreds to thousands of damage-induced ubiquitylation sites.
However, it is typically unknown which of the ~1000 ubiquitin ligases found in a human cell is responsible for an individual modification event, and which of these modification events are functionally important. In general, few attempts have thus been made to map ubiquitylation events catalysed by a specific ubiquitin ligase in a proteome-wide manner. We have now successfully achieved this for CSA, a ubiquitylation factor involved in TC-NER, and indirectly in RNAPII ubiquitylation.
In addition to identifying a number of interesting CSA target proteins and their specific ubiquitylation sites that we are presently pursuing, these techniques will be applied to other ubiquitin ligases, which are important in the DNA damage response.
Transcription during DNA damage
Bulky DNA lesions not only block the progress of RNAPII locally, but also affect transcription genome-wide so that even genes that are not damaged temporarily cease to be expressed.
The mechanisms and factors that underlie the more general, DNA damaged-induced repression of gene expression, and its eventual re-start, are still very poorly understood.
In order to uncover factors with a role in the transcription-related DNA damage response, we carried out several general and more specific proteomic screens, as well as a functional genomics screen.
The proteomic screens all made use of quantitative SILAC proteomics, enabling us to distinguish between ‘constitutive’ and UV-induced interactors of RNAPII and CSB, respectively. As well as the well-known TC-NER factors, this multi-omic approach has uncovered numerous new factors that had not previously been connected to the transcription-related DNA damage response, many of which we are presently investigating in detail.
Discovery of new factors via the multi-omic approach is complemented by genome-wide deep sequencing techniques such as ChIP-Seq, RNA-Seq, CLIP-Seq, 4SU-Seq and DRB/GRO-Seq to investigate the role of these new factors in the transcriptional response to UV-induced DNA damage at the genome-wide level.
Because UV-damage has turned out to trigger a dramatic change in mRNA splicing, termination, and RNA biology in general, much of our present focus has shifted more and towards RNA biology, including the function of stable RNAs.
- van den Heuvel, D., Rodríguez-Martínez, M., van der Meer, P.J., Nieto Moreno, N., Park, J., Kim, H.-S., van Schie, J.J.M., Wondergem, A.P., D'Souza, A., Yakoub, G., Herlihy, A.E., Kashyap, K., Boissière, T., Walker, J., Mitter, R., Apelt, K., de Lint, K., Kirdök, I., Ljungman, M., Wolthuis, R.M.F., Cramer, P., Schärer, O.D., Kokic, G.*, Svejstrup, J.Q.*, and Luijsterburg, M.S.* STK19 facilitates the clearance of lesion-stalled RNAPII during transcription-coupled DNA repair. (2024) Cell 187, 7107-7125.e25.
- Blears, D., Lou, J., Fong, N., Mitter, R., Sheridan, R.M., He, D., Dirac-Svejstrup, A.B., Bentley, D., and Svejstrup. J.Q. (2024). Redundant pathways for removal of defective RNA polymerase II complexes at a promoter-proximal pause checkpoint. Molecular Cell, 84, 4790-4807.e11.
- Han, Z., Moore, G.A., Mitter, R., Lopez Martinez, D., Wan, L., Dirac Svejstrup, A.B., Rueda, D.S. and Svejstrup, J.Q. (2023). DNA-directed termination of RNA Polymerase II transcription. Molecular Cell 83, 3253-3267.
- Cugusi, S., Mitter, R., Kelly, G.P., Walker, J., Han, Z., Pisano, P., Wierer, M., Stewart, A., and Svejstrup, J.Q. (2022). Heat shock induces premature transcript termination and reconfigures the human transcriptome. Molecular Cell 82, 1573-1588.e10.
- Wan, L., Juszkiewicz, S., Bajpe, P.K., Blears, D., Han, Z., Faull, P., Mitter, P., Snijders, A.P., Hegde, R.S., and Svejstrup, J.Q. (2021) cGAS Senses Translation Stress Through Direct Binding to Ribosomes. Molecular Cell 81, 2808-2822.e10.
- Dirac-Svejstrup, A.B., Walker, J., Faull, P., Encheva, V., Akimov, V., Puglia, M., Perkins, D. Kümper, S. Hunjan, S.S., Blagoev, B., Snijders, A.P., Powell, D.J., and Svejstrup, J.Q. (2020). DDI2 is a ubiquitin-directed endoprotease, responsible for cleavage of transcription factor NRF1. Molecular Cell 79, 332-341.E7.
- Rodríguez-Martínez, M., Boissiere, T., Noe Gonzalez, M., Litchfield, K., Mitter, R., Walker, J., Kjœr, S., Ismail, M., Downward, J., Swanton, C., and Svejstrup, J.Q. (2020). Evidence that STK19 is not a Melanoma Driver. Cell 181, 1395–1405.
- Tufegdzic Vidakovic, A., Mitter, R., Kelly, G., Neumann, M., Harreman, M., Rodriguez Martinez, M., Herlihy, A., Weems, J.C., Boeing, S., Encheva, V., Gaul, L., Milligan, L., Tollervey, D., Conaway, R.C., Conaway, J.W., Snijders, A.P., Stewart, A., and Svejstrup, J.Q. (2020). Regulation of the RNAPII Pool Is Integral to the DNA Damage Response. Cell 180, 1245-1261.
- Zatreanu, D., Han, Z., Mitter, R., Tumini, E., Williams, H., Gregersen, L.H., Dirac-Svejstrup, A.B., Roma, S., Stewart, A., Aguilera, A., and Svejstrup, J.Q. (2019) Elongation Factor TFIIS Prevents Transcription Stress and R-Loop Accumulation to Maintain Genome Stability. Molecular Cell 76, 57–69.
- Gregersen, L.H., Mitter, R., Ugalde, A.P., Nojima, T., Proudfoot, N. J., Agami, R., Stewart, A., and Svejstrup, J.Q. (2019). SCAF4 and SCAF8, mRNA Anti-Terminator Proteins. Cell 177, 1797–1813.
- Williamson, L., Saponaro, P., Boeing, S., East, P., Mitter, R., Kantidakis, T., Kelly, G.P., Lobley, A., Walker, J., Spencer-Dene, B., Howell, M., Stewart, A., and Svejstrup, J.Q. (2017). UV Irradiation Induces a Non-coding RNA that Functionally Opposes the Protein Encoded by the Same Gene. Cell 168, 843–855.
- Saponaro, M., Kantidakis, T., Mitter, R., Kelly, G.P., Heron, M., Williams, H., Söding, J., Stewart, A., and Svejstrup, J.Q. (2014). RECQL5 controls transcript elongation and suppresses genome instability associated with transcription stress. Cell 157, 1037-1049.

 Group Leader
Group Leader
Jesper Q. Svejstrup
Professor
Deputy head of department, Research
Director of CGEN
jsvejstrup@sund.ku.dk
(+45) 22 49 64 09
CV, publications, etc.
Transcription, RNA, and Gene Medicine Program
Center for Gene Expression
![]()
BRONZE 2023